Written informed consent was obtained from all participants, and the trial is being done in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. This study was approved in the UK by the Medicines and Healthcare products Regulatory Agency (reference 21584/0424/001-0001) and the South Central Berkshire Research Ethics Committee (reference 20/SC/0145). Vaccine use was authorised by Genetically Modified Organisms Safety Committees at each participating site.
This phase 1/2, participant-blinded, multicentre, randomised controlled trial is being done at five centres in the UK (Centre for Clinical Vaccinology and Tropical Medicine, University of Oxford; NIHR Southampton Clinical Research Facility, University Hospital Southampton NHS Foundation Trust, Southampton; Clinical Research Facility, Imperial College London; St Georges University of London and University Hospital NHS Foundation Trust; and University Hospitals Bristol and Weston NHS Foundation Trust). Healthy adult participants aged 18–55 years were recruited through local advertisements. All participants underwent a screening visit where a full medical history and examination was taken in addition to blood and urine tests (HIV; hepatitis B and C serology; full blood count; kidney and liver function tests; and urinary screen for blood, protein, and glucose and a pregnancy test done in women of childbearing potential). Volunteers with a history of laboratory confirmed SARS-CoV-2 infection; those at higher risk for SARS-CoV-2 exposure pre-enrolment (ie, front-line health-care workers working in emergency departments, intensive care units, and COVID-19 wards, and close contacts of confirmed COVID-19 cases; see appendix p 82 for further details); and those with a new onset of fever, cough, shortness of breath, and anosmia or ageusia since Feb 1, 2020, were excluded from the study. An amendment to the study protocol (amendment date April 21, 2020) allowed for recruitment of health-care workers with a negative SARS-CoV-2 serology at screening, once an antibody test became available. As it was not possible to screen for negative SARS-CoV-2 serology in all participants, some enrolled participants had high-level anti-spike antibodies at baseline and their data are included in all analyses. Full details of the eligibility criteria are described in the trial protocol provided in the appendix (pp 80–82)
Randomisation lists, using block randomisation stratified by study group and study site, were generated by the study statistician (MV). Block sizes of two and four were chosen to align with the study group sizes and the sequence of enrolment, and varied across study groups. Computer randomisation was done with full allocation concealment within the secure web platform used for the study electronic case report form (REDCap version 9.5.22; Vanderbilt University, Nashville, TN, USA). The trial staff administering the vaccine prepared vaccines out of sight of the participants and syringes were covered with an opaque material until ready for administration to ensure blinding of participants. Clinical investigators and the laboratory team remained blinded to group allocation.
Participants were randomly assigned (1:1) to receive either the ChAdOx1 nCoV-19 vaccine or the MenACWY vaccine. MenACWY was used as a comparator vaccine to maintain blinding of participants who experienced local or systemic reactions, since these reactions are a known association with viral vector vaccinations. Use of saline as a placebo would risk unblinding participants as those who had notable reactions would know they were in the ChAdOx1 nCoV-19 vaccine group.
et al. A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. 11 Morris SJ
Warimwe GM Laboratory-scale production of replication-deficient adenovirus vectored vaccines. 10 viral particles. The MenACWY vaccine was provided by the UK Department of Health and Social Care and administered as per summary of product characteristics at the standard dose of 0·5 mL. Vaccines were administered as a single intramuscular injection into the deltoid. The recombinant adenovirus for ChAdOx1 nCoV-19 was produced as previously described.The vaccine was manufactured according to current Good Manufacturing Practice by the Clinical BioManufacturing Facility (University of Oxford, Oxford, UK) as previously described,with only minor modifications, as described in the Investigational Medicinal Product Dossier and approved by the regulatory agency in the UK. ChAdOx1 nCoV-19 was administered at a dose of 5 × 10viral particles. The MenACWY vaccine was provided by the UK Department of Health and Social Care and administered as per summary of product characteristics at the standard dose of 0·5 mL. Vaccines were administered as a single intramuscular injection into the deltoid.
Participants were recruited and followed up according to groups. Participants were recruited first for groups 1 and 3, then group 2, and then group 4. Group 1 (the phase 1 component of the study) consisted of participants who had intensive early follow-up visits for safety and immunogenicity purposes at days 3, 7, 14, 28, and 56 after vaccination. Group 2 consisted of participants who had higher blood volumes drawn for humoral and cellular immunogenicity assessment than group 4, which consisted of participants who had a serum sample drawn for humoral immunology assessments only. Group 3 consisted of ten participants who were enrolled in a non-randomised prime-boost group and received a booster ChAdOx1 nCoV-19 administered 28 days after the first dose. These participants were not blinded and had extensive follow-up for safety and immunogenicity purposes, as per group 1, after each dose. A staggered-enrolment approach was used for the first two, six, and 90 participants recruited in groups 1 and 3 ( appendix p 89 ) and interim safety reviews with the independent Data and Safety Monitoring Board were done before proceeding with vaccinations in larger numbers of volunteers. Volunteers were considered enrolled into the trial at the point of vaccination.
Participants in all groups had blood samples drawn and clinical assessments for safety as well as immunology at days 0 and 28, and will also be followed up at days 184 and 364. A later amendment to the protocol (amendment date June 22, 2020) provided for additional testing of booster vaccinations in a subset of participants, the results of which are not yet available and are not included in this Article.
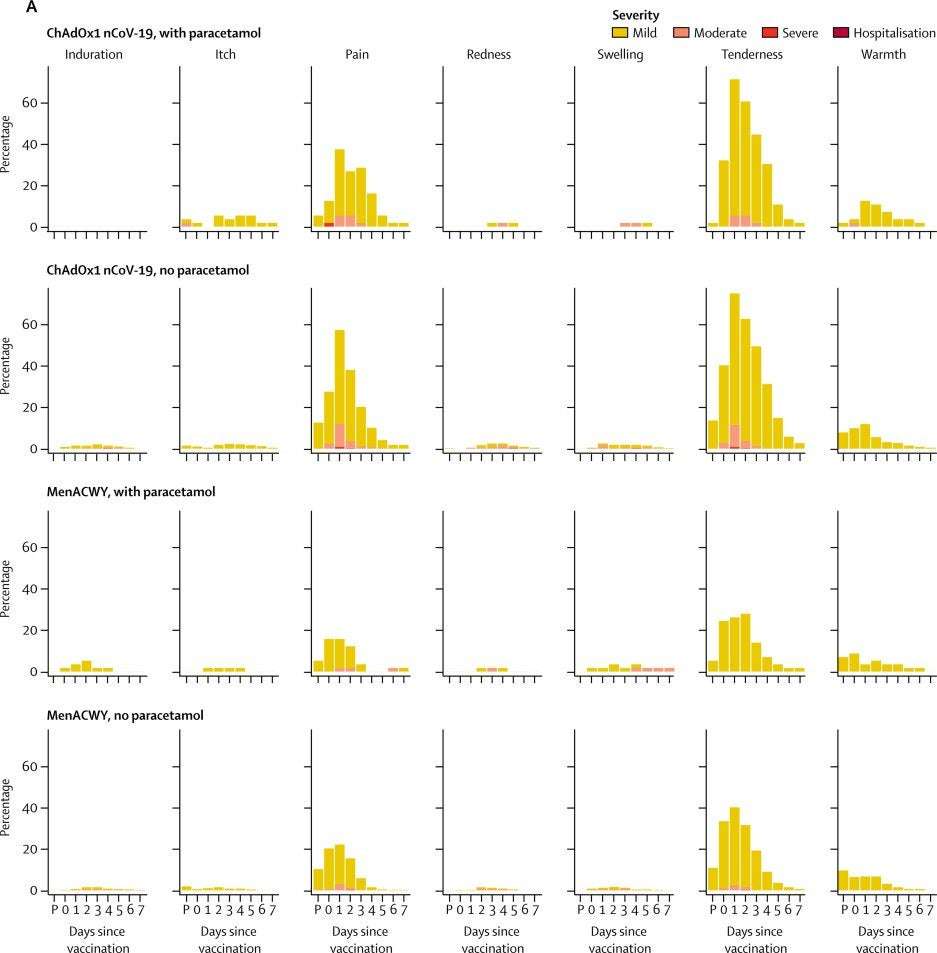
In two of the five trial sites (Oxford and Southampton), a protocol amendment (amendment date May 6, 2020) was implemented to allow prophylactic paracetamol to be administered before vaccination and participants were advised to continue with 1 g of paracetamol every 6 h for 24 h to reduce vaccine-associated reactions. All participants enrolled after the protocol amendment at these two sites were given prophylactic paracetamol and randomised equally to the vaccine or control arms of the study.
Participants were observed in the clinic for 30–60 min after the vaccination procedure and were asked to record any adverse events using electronic diaries during the 28-day follow-up period. Expected and protocol-defined local site reactions (injection site pain, tenderness, warmth, redness, swelling, induration, and itch) and systemic symptoms (malaise, muscle ache, joint pain, fatigue, nausea, headache, chills, feverishness [ie, a self-reported feeling of having a fever], and objective fever defined as an oral temperature of 38°C or higher) were recorded for 7 days. All other events were recorded for 28 days and serious adverse events are recorded throughout the follow-up period.
Severity of adverse events are graded with the following criteria: mild (transient or mild discomfort for <48 h, no interference with activity, and no medical intervention or therapy required), moderate (mild to moderate limitation in activity [some assistance might be needed] and no or minimal medical intervention or therapy required), severe (marked limitation in activity [some assistance usually required] and medical intervention or therapy required), and potentially life-threatening (requires assessment in emergency department or hospitalisation). Unsolicited adverse events are reviewed for causality by two clinicians blinded to group allocation, and events considered to be possibly, probably, or definitely related to the study vaccines were reported. Laboratory adverse events were graded by use of site-specific toxicity tables, which were adapted from the US Food and Drug Administration toxicity grading scale.
50 ], PHE microneutralisation assay [MNA IC 50 , IC 80 , IC 90 ], and Marburg virus neutralisation [VN IC 100 ]), and a pseudovirus neutralisation assay (PseudoNA IC 50 ). PHE PRNT is a live neutralisation assay and was done at PHE (Porton Down, UK). PHE MNA is a rapid microneutralisation assay, which was conducted in the same laboratory. The third assay, Marburg VN, was conducted at Marburg University (Marburg, Germany). Full details on the assays are provided in the Cellular responses were assessed using an ex-vivo interferon-γ enzyme-linked immunospot (ELISpot) assay to enumerate antigen-specific T cells. Humoral responses at baseline and following vaccination were assessed using a standardised total IgG ELISA against trimeric SARS CoV-2 spike protein, a muliplexed immunoassay (Meso Scale Discovery multiplexed immunoassay [MIA] against spike and receptor binding domain), three live SARS-CoV-2 neutralisation assays (Public Health England [PHE] plaque reduction neutralisation test [PRNT IC], PHE microneutralisation assay [MNA IC, IC, IC], and Marburg virus neutralisation [VN IC]), and a pseudovirus neutralisation assay (PseudoNA IC). PHE PRNT is a live neutralisation assay and was done at PHE (Porton Down, UK). PHE MNA is a rapid microneutralisation assay, which was conducted in the same laboratory. The third assay, Marburg VN, was conducted at Marburg University (Marburg, Germany). Full details on the assays are provided in the appendix (pp 31–34) . Owing to the labour-intensive nature of some of these assays, we prioritised analysis of samples from the ChAdOx1 nCoV-19 group, randomly selecting more samples from ChAdOx1 nCoV-19 participants than control samples to be sent for analysis.
Convalescent plasma samples from adults (≥18 years) with PCR-positive SARS-CoV-2 infection were obtained from symptomatic patients admitted to hospital or from surveillance on health-care workers who did not have symptomatic infection. These samples were tested using standardised ELISA, MIA, PseudoNA, and Marburg VN. Different samples were analysed across the assays, dependent on sample availability, laboratory capacity, and assay-specific requirements. Where multiple longitudinal samples were available for the same participant, only one timepoint is included in the analyses in this Article and the earliest timepoint (at least 20 days after initial symptoms) was selected.
Covered-in-Thorns on July 21st, 2020 at 02:07 UTC »
Is it 1,077, or 10,770?
saxy_sax_player on July 21st, 2020 at 01:46 UTC »
I have not read the article in full, but can anyone tell me why they haven’t tested adult above 55? Is that for a later phase because they’re higher risk?
beepybeetle on July 21st, 2020 at 01:22 UTC »
I found this Lancet article31605-6/fulltext) about the CanSino vaccine - which I read about alongside the Oxford one in the NYT - in “phase 2” trials as well, but what does phase 2 mean? do the vaccines need wider testing before it goes out to the broader public? pls explain to a dumb econ student