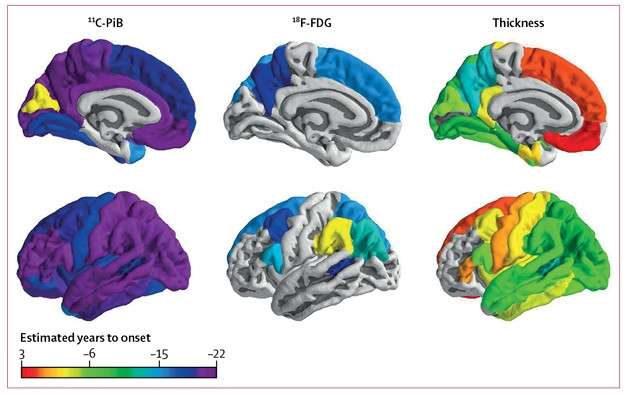
In the brains of carriers of familial Alzheimer’s disease mutations, Aβ starts accumulating in some regions more than two decades before symptoms appear, metabolism wanes six years later, and the brain begins to shrink about 10 years after that, or about five years prior to onset. This pattern plays out in many regions of the brain, according to the largest longitudinal neuroimaging study to date of people in the Dominantly Inherited Alzheimer Network (DIAN). Brian Gordon, Tyler Blazey, and colleagues at Washington University in St. Louis reported that this cascade emanated from the precuneus. However, some regions of the brain sidestepped this order. For example, their metabolism stayed normal despite Aβ build-up, or they shrank without accumulating plaques. Published in the March Lancet Neurology with dramatic videos of growing pathology, the findings paint a complex regional picture of neuropathological changes in preclinical AD.
DIAN tracked biomarkers starting 29 years before symptoms.
In general, Aβ accumulated first, then metabolism waned, then the cortex thinned, though some regions deviated from this sequence.
The precuneus succumbed first, accumulating Aβ more than 20 years before symptoms.
“The authors present some of the most reliable evidence to date for early biomarkers of Alzheimer’s disease more than 20 years before dementia onset,” wrote Arthur Toga of the University of Southern California in Los Angeles in a comment to Alzforum.
“This study supports the evidence that amyloid pathology drives neurodegenerative processes in early onset AD, observations that we have also reported in preclinical late-onset AD,” commented Heidi Jacobs of Massachusetts General Hospital in Boston. “[It] challenges us to look more closely at regional differences in the temporal time line of biomarkers,” she added.
In people with AD, memory problems reflect decades of pathological change brewing in the brain. DIAN comprises families who carry autosomal-dominant mutations in the PSEN1, PSEN2, or APP genes. Affected members develop early onset AD at an age researchers can predict based on family history and mutation. With the exception of small longitudinal studies, most published data on autosomal-dominant AD have been cross-sectional thus far, and support the idea that AD pathology initiates decades prior to estimated symptom onset and spreads from region to region throughout the brain (Jul 2012 news; Benzinger et al., 2013; Fleisher et al., 2015).
Working with Tammie Benzinger at WashU, Gordon, Blazey, and colleagues tapped into the ever-growing cache of longitudinal, neuroimaging data from DIAN to track pathological changes both regionally and temporally. They analyzed data from 229 mutation carriers and 148 noncarriers, whose age ranged from 29 years younger than expected age of onset to 10 years older. So far, more than half of these participants have been scanned at least twice, enabling longitudinal analysis. Those with serial data were scanned up to six times, averaging 2.4 scans per person. The researchers used PiB-PET to measure Aβ accumulation, FDG-PET to assess glucose metabolism, and structural MRI to measure thinning in 41 regions of the brain, 34 cortical and seven subcortical.
The researchers fed both serial and cross-sectional data into a Bayesian statistical model to calculate annual rates of change for each marker. For each region, the researchers estimated the earliest time in the disease course when rates of change for each imaging marker in carriers diverged from those in age-matched noncarriers.
Amyloid to Atrophy. The first signs of amyloid, hypometabolism, and cortical thinning by marker and by brain region. [Courtesy of Gordon et al., Lancet Neurology, 2018.]
“This study provides convincing evidence that the pattern of functional decline in autosomal-dominant Alzheimer's disease begins with Aβ deposition, progresses to metabolic decline, and ultimately culminates with structural decline before dementia symptoms arise—consistent with previous cross-sectional studies,” Toga wrote.
Aβ accumulation was the first of the three imaging measures to accelerate relative to noncarriers. This happened broadly across the brain. In 32 of 34 cortical regions, and in three of the subcortical regions, the annual change in PiB-PET uptake started to ramp up in mutation carriers 18.9 years before symptom onset on average. This acceleration occurred in the precuneus first, 22 years before onset. The posterior cingulate gyrus and medial orbital frontal cortex followed soon after, at 21 years. In the precuneus, and in the majority of other regions that developed Aβ pathology, the rate of deposition accelerated rapidly at first, then peaked but remained positive even after symptom onset. In a few regions of the brain, including the hippocampus and thalamus, the rate of PiB uptake never deviated significantly from that in noncarriers.
Aβ Accumulation. Annual rates of change (top) in PiB-PET uptake (bottom) mapped throughout the brain from 24 years prior to onset to 10 years after onset. Click to play movie. [Courtesy of Gordon et al., Lancet Neurology, 2018.]
At an average of 14.1 years before symptom onset, neuronal activity as measured by brain glucose metabolism started to slow down in carriers. This was much more focal than Aβ deposition, occurring in only eight cortical regions and no subcortical ones. Again, the precuneus was the canary in the coalmine, plunging into a metabolic downturn 18 years prior to symptoms. Glucose metabolism continued to plummet in this region until stabilizing about five years prior to clinical disease.
Metabolic Plunge. Annual rates of change (top) in FDG-PET uptake (bottom) mapped throughout the brain from 24 years prior to onset to 10 years after. Click to play movie. [Courtesy of Gordon et al., Lancet Neurology, 2018.]
Finally, at an average of 4.7 years prior to onset, parts of the brain started shriveling. Atrophy was widespread, ultimately affecting 24 of 34 cortical and all seven subcortical regions. The precuneus caved first, about 13 years prior to onset, while a few regions, such as the medial orbital frontal and superior frontal, evaded this structural expression of neurodegeneration until after clinical disease manifested.
For investigators who study biomarker changes in specific brain regions, the researchers created an online interface that displays the data and can be filtered in various ways (see DIAN longitudinal data).
Thinning Out Fast. Annual rates of change (top) in cortical thickness (bottom) mapped throughout the brain from 24 years prior to onset to 10 years after onset. Click to play movie. [Courtesy of Gordon et al., Lancet Neurology, 2018.]
Toga also drew attention to the significant regional and temporal differences in the cascade across the brain. “The considerable heterogeneity observed across regions suggests that vulnerability evolves both spatially and temporally as the disease progresses. This will be useful in guiding future biomarker research and in designing clinical trials.” The picture will become clearer as more longitudinal data roll in, Gordon said, as more participants have three or more visits.
One obvious missing piece of the puzzle is tau pathology. Many studies tie tau more closely to neurodegeneration than they do Aβ. A recent cross-sectional tau-PET imaging study in carriers of the PSEN1 E280A mutation saw tau accumulate in the medial temporal lobe a few years prior to symptom onset and spread into the cortex as memory problems began (Feb 2018 news). Preliminary tau PET data from the DIAN cohort largely agrees. Gordon told Alzforum that longitudinal tau imaging data from the cohort will soon be published, along with cognitive findings to tie the entire cascade together (Aug 2017 conference news).
Weaving tau PET into the picture could shed light on the regional heterogeneity in the pathological cascade reported here. For example, the hippocampus shrank despite evading Aβ pathology and metabolic dysfunction; this region is known to develop tau pathology early in AD. Gordon proposed that the hippocampus may actually harbor very low amounts of Aβ aggregates that are sufficient to stoke tau pathology and atrophy in the region.
Jacobs from MGH pointed out that the medial temporal lobe, which includes the hippocampus, is tightly connected to the precuneus and posterior cingulate cortex (PCC), two regions bearing the earliest burden of Aβ accumulation in the DIAN cohort. Jacobs recently reported that the erosion of white-matter tracts connecting the hippocampus to the PCC correlated with elevated tau pathology in the PCC and with memory problems (Feb 2018 news). “These connections may be related to the mechanisms underlying the interaction between amyloid and tau pathology in Alzheimer’s disease,” she wrote.
In a commentary accompanying Gordon’s paper, Betty Tijms and Pieter Jelle Visser and of VU University Medical Center in Amsterdam also proposed that Aβ aggregation might interact with tau via these same connections. Importantly, tau PET imaging in PSEN1 E280A carriers also showed early tau accumulation in the precuneus (Quiroz et al., 2018).
What renders the precuneus vulnerable to Aβ, and potentially tau, accumulation? Some researchers, most notably co-author Marcus Raichle of Washington University, favor the idea that its susceptibility stems from its unique metabolic characteristics, including a dependence on aerobic glycolysis, a glucose-burning pathway that plummets in the region with aging (Aug 2017 news; Sep 2010 news). Though Aβ deposition appeared long before the drop in glucose uptake in Gordon’s study, the researchers did not assess aerobic glycolysis specifically.
Whether the brain changes similarly in late-onset AD (LOAD) remains to be seen. Gordon pointed out that compared with the relatively young DIAN cohort, people with LOAD are likely to have additional metabolic, neuroinflammatory, and vascular confounders that could influence the temporal and regional progression of pathology in the brain.
Tijms and Visser noted that in most brain regions affected by hypometabolism in the DIAN cohort, this dysfunction closely trailed Aβ accumulation, and both occurred long before atrophy. However, in sporadic AD, hypometabolism has been more closely tied to atrophy (Nov 2017 news). “Hypometabolism in mutation carriers might result from mitochondrial dysfunction caused by specific mutations in APP and PSEN1, and in sporadic AD, hypometabolism might be more closely related to atrophy,” they proposed.
Yakeel Quiroz of Massachusetts General Hospital in Boston, who recently reported Aβ and tau accumulation in the precunei of PSEN1 E280A carriers, commended the authors. “They did an amazing job describing the temporal and spatial patterns of biomarker changes in preclinical AD, which has great relevance for clinical practice and clinical trials,” she wrote.—Jessica Shugart
TheGreening on February 24th, 2018 at 20:15 UTC »
Knowing the early symptoms, are there any postulations on how to prevent it from unfolding into the full blown disease?
helpinghat on February 24th, 2018 at 18:45 UTC »
What is Aβ and is there some way to prevent it from building up?
SirT6 on February 24th, 2018 at 18:44 UTC »
Timeline of familial Alzheimer’s Disease describes in this paper:
20 years before onset of symptoms: Amyloid starts to accumulate to detectable levels in the brain
14 years before onset of symptoms: Neurons in the brain begin to show signs of reduced metabolic activity.
5 years before onset of symptoms: The brain begins to atrophy.
What does this mean? What more do we need to know?
As the article suggests, understanding where Tau falls in this Alzheimer’s cascade is likely important. Other studies suggest fibriliary tangles emerge one to two years before disease onset.
How does this compare to age-matched controls (i.e. what does normal brain aging look like)?
How does this compare to sporadic AD (obviously a much harder study to do)?
Therapeutics? If amyloid is beginning to deposit decades before disease onset, is it reasonable to hope that amyloid targeting agents could work without being given for 10+ years in prophylaxis?